Attuazione delle direttive 2006/17/CE e 2006/86/CE, che attuano la direttiva 2004/23/CE per quanto riguarda le prescrizioni tecniche per la [...]
| Settore: | Normativa nazionale |
| Materia: | 86. Sanità |
| Capitolo: | 86.14 trapianti e donazioni di organi |
| Data: | 25/01/2010 |
| Numero: | 16 |
| Sommario |
| Art. 1. Campo d'applicazione |
| Art. 2. Definizioni |
| Art. 3. Approvvigionamento di tessuti e cellule umani |
| Art. 4. Criteri di selezione dei donatori |
| Art. 5. Esami di laboratorio |
| Art. 6. Donazione e approvvigionamento di tessuti e/o di cellule e ricevimento presso l'istituto dei tessuti |
| Art. 7. Distribuzione diretta ai riceventi di determinati tessuti e cellule |
| Art. 8. Prescrizioni per l'autorizzazione e l'accreditamento degli Istituti dei tessuti |
| Art. 9. Prescrizioni per l'autorizzazione e l'accreditamento allo svolgimento dei procedimenti di preparazione di tessuti e cellule |
| Art. 10. Notifica di reazioni avverse gravi |
| Art. 11. Notifica di eventi avversi gravi |
| Art. 12. Relazioni annuali |
| Art. 13. Scambio di informazioni fra Autorità competenti |
| Art. 14. Rintracciabilità |
| Art. 15. (Sistema di codifica europeo). |
| Art. 15 bis. (Formato del codice unico europeo). |
| Art. 15 ter. (Prescrizioni relative all'applicazione del codice unico europeo). |
| Art. 16. Recepimento |
| Art. 16 bis. (Sospensione o revoca dell'autorizzazione e dell'accreditamento degli Istituti dei tessuti). |
| Art. 17. Clausola di cedevolezza |
| Art. 18. Disposizioni finanziarie |
§ 86.14.41 - D.Lgs. 25 gennaio 2010, n. 16.
Attuazione delle direttive 2006/17/CE e 2006/86/CE, che attuano la direttiva 2004/23/CE per quanto riguarda le prescrizioni tecniche per la donazione, l'approvvigionamento e il controllo di tessuti e cellule umani, nonchè per quanto riguarda le prescrizioni in tema di rintracciabilità, la notifica di reazioni ed eventi avversi gravi e determinate prescrizioni tecniche per la codifica, la lavorazione, la conservazione, lo stoccaggio e la distribuzione di tessuti e cellule umani.
(G.U. 18 febbraio 2010, n. 40)
IL PRESIDENTE DELLA REPUBBLICA
Visti gli articoli 76 e 87 della Costituzione;
Vista la
Visto il
Vista la
Vista la
Vista la
Vista la legge 1° aprile 1999, n. 91, recante disposizioni in materia di trapianti di organi e di tessuti;
Vista la
Vista la
Visto il decreto del Presidente della Repubblica 14 gennaio 1997, pubblicato nella Gazzetta Ufficiale n. 42 del 20 febbraio 1997, recante approvazione dell'atto di indirizzo e coordinamento alle regioni e alle province autonome di Trento e di Bolzano, in materia di requisiti strutturali, tecnologici ed organizzativi minimi per l'esercizio delle attività sanitarie da parte delle strutture pubbliche e private;
Visto il
Visto il decreto del Presidente del Consiglio dei Ministri in data 1° settembre 2000, pubblicato nella Gazzetta Ufficiale n. 274 del 23 novembre 2000, recante approvazione dell'atto di indirizzo e coordinamento in materia di requisiti strutturali, tecnologici ed organizzativi minimi per l'esercizio delle attività sanitarie relative alla medicina trasfusionale;
Vista la
Visto il
Visto il
Visto l'Accordo 21 marzo 2002 tra il Ministero della salute, le regioni e le province autonome di Trento e di Bolzano, concernente linee guida per le attività di coordinamento per il reperimento di organi e di tessuti in ambito nazionale ai fini di trapianto;
Visto l'Accordo 21 marzo 2002 tra il Ministero della salute, le regioni e le province autonome di Trento e di Bolzano, concernente linee guida per il prelievo, la conservazione e l'utilizzo di tessuto muscolo-scheletrico;
Visto il decreto del Ministro della salute in data 2 dicembre 2004, pubblicato nella Gazzetta Ufficiale n. 27 del 3 febbraio 2005, recante modalità per il rilascio delle autorizzazioni all'esportazione o all'importazione di organi e tessuti;
Vista la
Visto il decreto del Ministro della salute in data 11 aprile 2008, pubblicato nella Gazzetta Ufficiale n. 101 del 30 aprile 2008, recante linee guida in materia di procreazione medicalmente assistita;
Visto l'Accordo 23 settembre 2004 tra il Ministero della salute, le regioni e le province autonome di Trento e di Bolzano, sul documento recante linee guida sulle modalità di disciplina delle attività di reperimento, trattamento, conservazione e distribuzione di cellule e tessuti umani a scopo, in attuazione dell'articolo 15, comma 1, della legge 1° aprile 1999, n. 91;
Visto l'Accordo 10 luglio 2003 tra il Ministero della salute, le regioni e le province autonome di Trento e di Bolzano, sul documento recante linee guida in tema di raccolta, manipolazione e impiego clinico delle cellule staminali emopoietiche (CSE);
Visto l'Accordo 5 ottobre 2006 tra il Ministero della salute, le regioni e le province autonome di Trento e di Bolzano in materia di ricerca e reperimento di cellule staminali emopoietiche presso registri e banche italiane ed estere;
Visto il
Vista la preliminare deliberazione del Consiglio dei Ministri, adottata nella riunione del 2 ottobre 2009;
Acquisito il parere della Conferenza permanente per i rapporti tra lo Stato, le regioni e le province autonome di Trento e di Bolzano nella seduta del 29 ottobre 2009;
Acquisiti i pareri delle competenti Commissioni della Camera dei deputati e del Senato della Repubblica;
Vista la deliberazione del Consiglio dei Ministri, adottata nella riunione del 13 gennaio 2010;
Sulla proposta del Ministro per le politiche europee e del Ministro della salute, di concerto con i Ministri degli affari esteri, della giustizia, dell'economia e delle finanze e per i rapporti con le regioni;
Emana
il seguente decreto legislativo:
Art. 1. Campo d'applicazione
1. Il presente decreto disciplina determinate prescrizioni tecniche per la donazione, l'approvvigionamento e il controllo di tessuti e cellule umani, nonchè la codifica, la lavorazione, la conservazione, lo stoccaggio e la distribuzione di:
a) tessuti e cellule umani, destinati ad applicazioni sull'uomo;
b) prodotti fabbricati, derivati da tessuti e cellule umani destinati ad applicazioni sull'uomo, qualora tali prodotti non siano disciplinati da altre direttive.
2. Le disposizioni degli articoli da 10 a 14 del presente decreto, relative alla rintracciabilità e alla notifica di reazioni ed eventi avversi gravi, si applicano anche alla donazione, all'approvvigionamento e al controllo di tessuti e cellule umani.
3. Ai fini dell'attuazione delle disposizioni recate in materia di cellule riproduttive dal
Art. 2. Definizioni
1. Ai fini del presente decreto si intendono per:
a) cellule riproduttive: tutti i tessuti e le cellule destinati ad essere utilizzati ai fini della riproduzione assistita, nel rispetto delle disposizioni vigenti in materia di procreazione medicalmente assistita;
b) donazione da parte di un partner: la donazione di cellule riproduttive tra un uomo e una donna che rispettino i requisiti soggettivi di cui all'articolo 5 della
c) impiego diretto: qualsiasi procedura in base alla quale le cellule donate vengono utilizzate senza essere conservate;
d) sistema di qualità: la struttura organizzativa, le responsabilità, le procedure, i processi e le risorse destinati ad attuare la gestione della qualità, comprese tutte le attività che direttamente o indirettamente contribuiscono alla qualità;
e) gestione della qualità: le attività coordinate per dirigere e controllare un'organizzazione sul piano della qualità;
f) procedure operative standard (POS): istruzioni scritte che descrivono le fasi di un determinato processo nonchè i materiali e i metodi da utilizzare e il prodotto finale previsto;
g) convalida, o qualifica in caso di attrezzature o ambienti: la produzione di prove documentate, in grado di garantire con un elevato livello di certezza che determinati procedimenti, attrezzature o ambienti diano luogo a un prodotto conforme alle specifiche e alle caratteristiche qualitative prestabilite; un procedimento è convalidato al fine di valutare se il sistema funziona efficacemente in rapporto all'impiego previsto;
h) rintracciabilità: la possibilità di ricostruire il percorso di tessuti o cellule in ogni fase dell'approvvigionamento, della lavorazione, del controllo e dello stoccaggio fino alla distribuzione al ricevente o allo smaltimento, compresa la possibilità di risalire all'identificazione del donatore, dell'istituto dei tessuti o del centro di produzione che ricevono, o lavorano o stoccano i tessuti o le cellule, nonchè, a livello delle strutture sanitarie, la possibilità di individuare i responsabili che applicano i tessuti o le cellule sui riceventi. Tale rintracciabilità riguarda anche la possibilità di reperire e identificare tutti i dati pertinenti relativi ai prodotti e ai materiali che vengono a contatto con detti tessuti o cellule;
i) critico: che ha potenzialmente effetto sulla qualità e o la sicurezza di tessuti e cellule o è a contatto con i predetti;
l) organizzazione per l'approvvigionamento: una struttura sanitaria o un'unità ospedaliera in cui si effettuano prelievi di tessuti e cellule umani, che può non essere autorizzata e accreditata come Istituto dei tessuti, fatto salvo quanto previsto dalla
m) organizzazione responsabile dell'applicazione sull'uomo: una struttura sanitaria o un'unità ospedaliera che esegue applicazioni sull'uomo di tessuti e cellule;
m-bis) "codice unico europeo" o Single European Code (SEC): il codice unico d'identificazione applicato ai tessuti e alle cellule distribuiti nell'Unione; il codice unico europeo è costituito da una sequenza d'identificazione della donazione e da una sequenza d'identificazione del prodotto, secondo quanto specificato nell'allegato XI del presente decreto [2];
m-ter) "sequenza d'identificazione della donazione (SID)": la prima parte del codice unico europeo costituita dal codice dell'istituto dei tessuti dell'Unione europea (UE) e dal numero unico della donazione [3];
m-quater) "codice dell'istituto dei tessuti dell'UE": il codice unico d'identificazione degli istituti dei tessuti autorizzati e accreditati, ai sensi degli articoli 6 e 7 del
m-quinquies) "numero unico della donazione": il numero unico attribuito a una determinata donazione di tessuti o di cellule conformemente al sistema in vigore in uno Stato membro per l'assegnazione di tali numeri, che in Italia è rappresentato dal numero unico nazionale attribuito dal Sistema Informativo Trapianti (SIT), secondo quanto specificato nell'allegato XI del presente decreto [5];
m-sexies) "sequenza d'identificazione del prodotto (SIP)": la seconda parte del codice unico europeo costituita dal codice del prodotto, dal numero specifico della sottopartita e dalla data di scadenza [6];
m-septies) "codice del prodotto": il codice d'identificazione per il tipo specifico di tessuti e di cellule in questione; il codice del prodotto è costituito dal codice d'identificazione del sistema di codifica del prodotto indicante il sistema di codifica utilizzato dall'istituto dei tessuti ("E" per EUTC, "A" per ISBT128, "B" per Eurocode) e il codice della tipologia di prodotto di tessuti e cellule previsto nel rispettivo sistema di codifica per il tipo di prodotto, secondo quanto specificato nell'allegato XI del presente decreto [7];
m-octies) "numero specifico della sottopartita": il numero che distingue le aliquote di tessuti e cellule e che identifica in maniera univoca i tessuti e le cellule aventi lo stesso numero unico della donazione e lo stesso codice del prodotto e provenienti dallo stesso istituto dei tessuti, secondo quanto specificato nell'allegato XI del presente decreto [8];
m-novies) "data di scadenza": la data entro la quale i tessuti e le cellule possono essere applicati, secondo il formato specificato nell'allegato XI del presente decreto [9];
m-decies) "piattaforma di codifica dell'UE": la piattaforma informatica ospitata dalla Commissione che contiene il compendio degli istituti dei tessuti dell'UE e il compendio dei prodotti di tessuti e cellule dell'UE [10];
m-undecies) "compendio degli istituti dei tessuti dell'UE": il registro di tutti gli istituti dei tessuti titolari di licenza, autorizzati, designati o accreditati dall'autorità competente o dalle autorità competenti degli Stati membri, inclusi gli istituti autorizzati ed accreditati dalle regioni e province autonome, ai sensi degli articoli 6 e 7 del
m-duodecies) "compendio dei prodotti di tessuti e cellule dell'UE": il registro di tutti i tipi di tessuti e di cellule che circolano nell'Unione e i rispettivi codici del prodotto nell'ambito dei tre sistemi di codifica autorizzati (EUTC, ISBT128 ed Eurocode) [12];
m-terdecies) "EUTC": il sistema di codifica del prodotto per i tessuti e le cellule sviluppato dall'Unione e costituito da un registro di tutti i tipi di tessuti e di cellule che circolano nell'Unione e dai corrispondenti codici del prodotto [13];
m-quaterdecies) "rilascio per la circolazione": la distribuzione a fini di applicazioni sull'uomo o il trasferimento a un altro Istituto dei tessuti o altra parte terza, ad esempio per l'ulteriore lavorazione con o senza restituzione [14];
m-quinquiesdecies) "all'interno dello stesso centro": tutte le fasi, dall'approvvigionamento all'applicazione sull'uomo, sono svolte sotto la responsabilità di una stessa persona, applicando i medesimi sistemi di gestione della qualità e di rintracciabilità, nell'ambito di una struttura sanitaria comprendente almeno un istituto dei tessuti autorizzato e accreditato, e un'organizzazione responsabile delle applicazioni sull'uomo nella stessa sede [15];
m-sexiesdecies) "pooling": il contatto fisico o la mescolanza in un singolo contenitore di tessuti o di cellule provenienti da più di un approvvigionamento dallo stesso donatore o da due o più donatori [16];
m-septiesdecies) "Sistema Informativo Trapianti" (di seguito nominato "SIT"), sistema informativo di supporto per l'informatizzazione delle attività della Rete nazionale trapianti, di cui all'articolo 7, comma 2, della legge 1° aprile 1999, n. 91 [17];
m-octiesdecies) "ISBT 128": standard internazionale per la terminologia, l'identificazione, la codifica e l'etichettatura dei prodotti di origine umana come definiti dall'Organizzazione mondiale della sanità (inclusi sangue, cellule, tessuti, latte e organi) e gestito da ICCBBA [18];
m-noviesdecies) "Eurocode": standard internazionale senza scopo di lucro per l'etichettatura dei prodotti del sangue e tessuti per il miglioramento della trasfusione di sangue e il trapianto di tessuti, gestito da Eurocode - Sistemi Internazionali di etichettatura [19].
Art. 3. Approvvigionamento di tessuti e cellule umani
1. Ad eccezione della donazione da parte di un partner di cellule riproduttive destinate all'impiego diretto, l'approvvigionamento di tessuti e cellule umani è autorizzato solo qualora siano rispettate le prescrizioni di cui ai commi da 2 a 12.
2. Il prelievo di tessuti e cellule umani è effettuato da personale qualificato e adeguatamente formato, ai sensi della normativa vigente, a svolgere tali attività.
3. L'istituto dei tessuti o l'organizzazione per l'approvvigionamento conclude con il personale qualificato o l'equipe clinica responsabile della selezione del donatore che non faccia parte dello stesso istituto o organizzazione, accordi scritti in ordine alle procedure da seguire per garantire la conformità dei requisiti ai criteri di selezione dei donatori di cui all'allegato I.
4. L'istituto dei tessuti o l'organizzazione per l'approvvigionamento conclude con il personale qualificato o l'equipe clinica responsabile della selezione del donatore che non faccia parte dello stesso istituto o organizzazione, accordi scritti in ordine al tipo di tessuti, di cellule o di campioni da prelevare nonchè ai protocolli da seguire.
5. Il responsabile dell'istituto dei tessuti, in accordo con l'organizzazione per l'approvvigionamento definisce procedure operative standard, in seguito indicate come «POS», al fine di verificare:
a) l'identità del donatore;
b) la documentazione relativa al consenso informato, o all'espressione di volontà o all'autorizzazione alla donazione da parte del donatore o della sua famiglia;
c) la valutazione dei criteri di selezione dei donatori di cui all'articolo 4;
d) la valutazione degli esami di laboratorio richiesti per i donatori di cui all'articolo 5.
6. Il responsabile dell'istituto dei tessuti, in accordo con l'organizzazione per l'approvvigionamento definisce altresì POS relative all'approvvigionamento, confezionamento, etichettatura e trasporto dei tessuti e delle cellule fino alla destinazione presso l'istituto dei tessuti o, in caso di distribuzione diretta di tali materiali, presso l'equipe clinica responsabile della loro applicazione, ovvero, in caso di campioni di tessuti e cellule, presso il laboratorio per il controllo, in conformità all'articolo 5.
7. L'approvvigionamento è eseguito presso strutture adeguate, ponendo in atto procedure mirate a ridurre il rischio di contaminazione batterica o di altro tipo dei tessuti e delle cellule prelevati ai sensi dell'articolo 6.
8. I materiali e le attrezzature utilizzati per l'approvvigionamento sono gestiti conformemente alle norme e alle specifiche di cui all'allegato IV, sezione 1.3, tenendo debitamente conto delle regolamentazioni, normative e linee guida nazionali ed internazionali, relative alla sterilizzazione di medicinali e dispositivi medici. Per il prelievo di tessuti e cellule sono impiegati appositi strumenti e dispositivi qualificati, sterili.
9. Il prelievo di tessuti e cellule da donatore vivente è effettuato in un contesto che ne garantisca la salute, la sicurezza e la tutela dei dati personali.
10. Nel caso di donatore cadavere è assicurata la disponibilità di personale e attrezzature per la ricomposizione del corpo che deve essere effettuata in modo completo ed efficace.
11. Le procedure relative al prelievo di tessuti e cellule sono attuate in conformità alle disposizioni di cui all'articolo 6.
12. Nel corso del prelievo o presso l'istituto dei tessuti viene assegnato un codice di identificazione unico al donatore ed ai tessuti e alle cellule donati, in modo da garantire un'adeguata identificazione del donatore e la tracciabilità dei materiali donati, nel rispetto delle norme per la tutela della riservatezza. I codici e i dati correlati sono annotati in un registro predisposto a tale fine. La codifica di cui al presente comma si uniforma a quella nazionale o europea di cui all'articolo 15 [20].
13. La documentazione relativa al donatore è conservata conformemente a quanto previsto dall'allegato IV, punto 1.4.
Art. 4. Criteri di selezione dei donatori
1. Il responsabile dell'organizzazione per l'approvvigionamento o dell'istituto dei tessuti garantisce che la selezione dei donatori sia effettuata in conformità ai criteri di selezione di cui:
a) all'allegato I per i donatori di tessuti e cellule;
b) all'allegato III per i donatori di cellule riproduttive.
2. Per i donatori di cellule staminali emopoietiche midollari e periferiche, ivi incluse quelle da sangue cordonale, si applicano anche le disposizioni previste dalla normativa vigente in materia di attività trasfusionali e di trapianto di midollo.
Art. 5. Esami di laboratorio
1. Il responsabile dell'organizzazione per l'approvvigionamento o dell'istituto dei tessuti garantisce che:
a) i donatori di tessuti e di cellule, ad eccezione dei donatori di cellule riproduttive, vengano sottoposti agli esami di cui all'allegato II, punto 1;
b) tali esami siano effettuati secondo le prescrizioni stabilite nell'allegato II, punto 2.
2. Il responsabile dell'organizzazione per l'approvvigionamento o dell'istituto dei tessuti garantisce che:
a) i donatori di cellule riproduttive siano sottoposti agli esami di cui all'allegato III, punti 1 e 2;
b) gli esami di cui alla lettera a) siano effettuati secondo le prescrizioni stabilite nell'allegato III, punto 3.
3. Per i donatori di cellule staminali emopoietiche periferiche, ivi incluse quelle da sangue cordonale, si applicano anche le disposizioni previste dalla normativa vigente in materia di attività trasfusionali.
Art. 6. Donazione e approvvigionamento di tessuti e/o di cellule e ricevimento presso l'istituto dei tessuti
1. Il responsabile dell'organizzazione per l'approvvigionamento o dell'istituto dei tessuti garantisce la conformità alle prescrizioni di cui all'allegato IV delle procedure relative alla donazione e all'approvvigionamento di tessuti o cellule nonchè al ricevimento degli stessi presso l'istituto dei tessuti.
Art. 7. Distribuzione diretta ai riceventi di determinati tessuti e cellule
1. Il responsabile dell'organizzazione per l'approvvigionamento o dell'istituto dei tessuti, acquisito il parere del Centro nazionale trapianti, può autorizzare la distribuzione diretta di determinati tessuti e cellule dal luogo in cui è effettuato il prelievo a un centro di assistenza sanitaria ai fini di un trapianto immediato.
Art. 8. Prescrizioni per l'autorizzazione e l'accreditamento degli Istituti dei tessuti
1. L'allegato V al presente decreto riporta le indicazioni e le prescrizioni da soddisfare ai fini del rilascio, da parte dell'Autorità regionale competente, dell'autorizzazione e dell'accreditamento.
Art. 9. Prescrizioni per l'autorizzazione e l'accreditamento allo svolgimento dei procedimenti di preparazione di tessuti e cellule
1. L'allegato VI al presente decreto riporta le indicazioni e le prescrizioni da soddisfare ai fini del rilascio, da parte dell'Autorità regionale competente, dell'autorizzazione allo svolgimento dei procedimenti di preparazione di tessuti e cellule.
Art. 10. Notifica di reazioni avverse gravi
1. In conformità ai disposti di cui all'articolo 11 del
a) l'organizzazione per l'approvvigionamento predispone procedure per conservare le registrazioni dei dati relativi a tessuti e cellule prelevati e per notificare tempestivamente all'istituto dei tessuti di riferimento ogni reazione avversa grave nel donatore vivente che possa influire sulla qualità e sicurezza di tessuti e cellule;
b) l'organizzazione responsabile dell'applicazione sull'uomo di tessuti e cellule predispone procedure per conservare le registrazioni dei dati di tessuti e cellule applicati sull'uomo e per notificare tempestivamente all'istituto dei tessuti di riferimento ogni reazione avversa grave osservata nel corso o a seguito dell'applicazione clinica, che possa essere in rapporto con la qualità e la sicurezza dei tessuti e delle cellule utilizzate;
c) l'istituto dei tessuti che distribuisce tessuti e cellule per applicazioni sull'uomo fornisce all'organizzazione responsabile dell'applicazione sull'uomo di tessuti e cellule, coerentemente a quanto previsto alla lettera b), informazioni sulle modalità per la notifica delle reazioni avverse gravi.
2. In conformità ai disposti di cui all'articolo 11 del
a) predispone procedure per comunicare tempestivamente alla rispettiva autorità regionale e al CNT o al Centro nazionale sangue, in seguito indicato come «CNS», secondo i rispettivi ambiti di competenza, tutte le informazioni disponibili attinenti alle presunte reazioni avverse gravi di cui al comma 1, lettere a) e b) [21];
b) predispone procedure per comunicare tempestivamente alla rispettiva autorità regionale e al CNT o al CNS, secondo i rispettivi ambiti di competenza, le conclusioni dell'indagine per analizzare le cause e il conseguente esito [22].
2-bis. Nel caso di cellule riproduttive o embrioni il CNT deve trasmettere tempestivamente e comunque non oltre 48 ore le informazioni ricevute di cui al comma 2, lettere a) e b), al Registro dell'ISS di cui all'articolo 11 della
3. Conformemente ai disposti di cui all'articolo 11 del
a) la persona responsabile di cui all'articolo 17 dello stesso decreto legislativo, comunica alla rispettiva autorità regionale e al CNT o al CNS, secondo i rispettivi ambiti di competenza, le informazioni incluse nel modello di notifica di cui alla parte A dell'allegato VII;
b) l'istituto dei tessuti notifica alla rispettiva Autorità regionale e al CNT o al CNS, secondo l'ambito di competenza, i provvedimenti adottati per quanto riguarda altri tessuti e cellule interessati, distribuiti a fini di applicazioni sull'uomo;
c) l'istituto dei tessuti notifica alla rispettiva Autorità regionale e al CNT o al CNS, secondo l'ambito di competenza, le conclusioni dell'indagine, fornendo almeno le informazioni di cui alla parte B dell'allegato VII.
Art. 11. Notifica di eventi avversi gravi
1. In conformità ai disposti di cui all'articolo 11 del
a) l'organizzazione per l'approvvigionamento e l'istituto dei tessuti predispongono procedure per conservare le registrazioni dei dati e per notificare tempestivamente ogni evento avverso grave che si verifichi durante l'approvvigionamento e possa influire sulla qualità e sicurezza dei tessuti e cellule;
b) l'organizzazione responsabile dell'applicazione sull'uomo di tessuti e cellule predispone procedure per notificare tempestivamente all'istituto dei tessuti di riferimento ogni evento avverso grave che possa influire sulla qualità e sicurezza dei tessuti e cellule;
c) l'istituto dei tessuti fornisce all'organizzazione responsabile dell'applicazione sull'uomo informazioni sulle modalità per notificargli eventi avversi gravi che possano influire sulla qualità e sicurezza dei tessuti e cellule.
2. In materia di riproduzione assistita si considera evento avverso grave ogni tipo di errore d'identificazione o di scambio di gameti o embrioni. La persona interessata o l'organizzazione per l'approvvigionamento o l'organizzazione responsabile dell'applicazione sull'uomo riferiscono tali eventi all'istituto dei tessuti fornitore ai fini dell'indagine e notifica all'autorità competente.
3. In conformità ai disposti di cui all'articolo 11 del
a) l'istituto dei tessuti predispone procedure per comunicare tempestivamente alla rispettiva autorità regionale e al CNT o al CNS, secondo l'ambito di competenza, tutte le informazioni disponibili attinenti ai presunti eventi avversi gravi di cui al comma 1, lettere a) e b) [24];
b) l'istituto dei tessuti o le autorità regionali valutano e comunicano al CNT o CNS, secondo l'ambito di competenza, l'eventuale implicazione di altre cellule e tessuti, e gli eventuali provvedimenti intrapresi [25];
c) l'istituto dei tessuti predispone procedure per comunicare tempestivamente alla rispettiva autorità regionale e al CNT o al CNS, secondo l'ambito di competenza, le conclusioni dell'indagine per analizzare le cause e il conseguente esito [26].
3-bis. Nel caso di cellule riproduttive o embrioni il CNT deve trasmettere tempestivamente e comunque non oltre 48 ore le informazioni ricevute di cui al comma 3, lettere a), b) e c), al Registro dell'ISS, di cui all'articolo 11 della
4. In conformità ai disposti di cui all'articolo 11 del
a) il responsabile dell'istituto dei tessuti notifica alla rispettiva autorità regionale e al CNT o al CNS, secondo l'ambito di competenza, le informazioni incluse nel modello di notifica di cui alla parte A dell'allegato VIII;
b) l'istituto dei tessuti valuta gli eventi avversi gravi per individuarne le cause evitabili nell'ambito del procedimento;
c) l'istituto dei tessuti notifica alla rispettiva autorità regionale e al CNT o al CNS, secondo l'ambito di competenza, le conclusioni dell'indagine, fornendo almeno le informazioni di cui alla parte B dell'allegato VIII.
Art. 12. Relazioni annuali
1. Il Ministero della salute entro il 30 giugno di ogni anno presenta alla Commissione una relazione annuale sulle notifiche delle reazioni e degli eventi avversi gravi, relative all'anno precedente, ricevute dalle Autorità competenti. Il Ministero della salute mette tale relazione a disposizione degli istituti dei tessuti.
2. La trasmissione dei dati si attiene alle specifiche del modello di scambio di dati di cui all'allegato IX, parti A e B, e fornisce tutte le informazioni necessarie ad identificare il mittente e a conservare i suoi dati di riferimento.
Art. 13. Scambio di informazioni fra Autorità competenti
1. Il Ministero della salute pone in atto le iniziative necessarie ad assicurare la comunicazione delle informazioni del caso ai rispettivi organismi degli Stati membri in relazione a reazioni ed eventi avversi gravi, al fine di assicurare l'adozione dei necessari adeguati provvedimenti.
Art. 14. Rintracciabilità [28]
1. L'Istituto dei tessuti garantisce che i tessuti e le cellule siano rintracciabili, in particolare mediante la documentazione e l'uso del codice unico europeo, dall'approvvigionamento all'applicazione sull'uomo o allo smaltimento e viceversa. I tessuti e le cellule utilizzati per i medicinali per terapie avanzate sono rintracciabili ai sensi del presente decreto almeno fino al loro trasferimento a produttori di tali medicinali.
2. L'Istituto dei tessuti e l'organizzazione responsabile delle applicazioni sull'uomo conservano per almeno 30 anni i dati di cui all'allegato X, avvalendosi di un sistema di archiviazione appropriato e leggibile.
3. Nel caso di tessuti e di cellule prelevati da un donatore deceduto da parte di equipe di prelievo operanti per due o più istituti dei tessuti, viene garantito, attraverso il SIT, un appropriato sistema di rintracciabilità per tutti i tessuti e le cellule prelevati, tramite l'assegnazione della SID.
Art. 15. (Sistema di codifica europeo). [29]
1. Fatto salvo il comma 2 del presente articolo, si applica un codice unico europeo a tutti i tessuti e a tutte le cellule, comprese le cellule staminali emopoietiche, distribuiti a fini di applicazioni sull'uomo. Negli altri casi in cui i tessuti e le cellule sono rilasciati per la circolazione, è applicata come minimo la SID almeno nei documenti di accompagnamento.
2. Il comma 1 non si applica:
a) alla donazione di cellule riproduttive dal partner;
b) ai tessuti e alle cellule distribuiti direttamente per il trapianto immediato al ricevente, ai sensi dell'articolo 6, comma 6, del
c) ai tessuti e alle cellule importati in Italia in caso di emergenza allorchè l'importazione è autorizzata direttamente dall'autorità competente, ai sensi dell'articolo 9, comma 3, lettera b), del
Art. 15 bis. (Formato del codice unico europeo). [30]
1. Il codice unico europeo di cui all'articolo 15, comma 1, è conforme alle specifiche di cui al presente articolo e all'allegato XI.
2. Il codice unico europeo, in formato leggibile all'occhio umano, è preceduto dalla sigla "SEC". È possibile l'uso parallelo di altri sistemi di etichettatura e rintracciabilità.
3. Il codice unico europeo deve riportare le sequenze di identificazione SID e SIP separate da un unico spazio o come due righe successive.
Art. 15 ter. (Prescrizioni relative all'applicazione del codice unico europeo). [31]
1. Gli istituti dei tessuti, compresi gli istituti dei tessuti importatori (ITI), quali definiti dalla
a) assegnare il codice unico europeo, costituito dalla SID, generata dal SIT, e dalla SIP, assegnata mediante sistemi locali degli Istituiti dei tessuti, a tutti i tessuti e a tutte le cellule per i quali è richiesta l'applicazione di tale codice, al più tardi prima della loro distribuzione a fini di applicazioni sull'uomo;
b) assegnare la SID, generata dal SIT, dopo l'approvvigionamento dei tessuti e delle cellule, o al momento del loro ricevimento da un'organizzazione di approvvigionamento o all'atto dell'importazione di tessuti e cellule da un Paese terzo; la SID comprende:
1) il pertinente codice dell'istituto dei tessuti italiano quale assegnato nel compendio degli istituti dei tessuti dell'UE,
2) il numero unico della donazione attribuito a livello nazionale dal SIT; in caso di pooling di tessuti e cellule, al prodotto finale è assegnato un nuovo numero d'identificazione della donazione; la rintracciabilità delle singole donazioni è assicurata dall'istituto dei tessuti in cui è effettuato il pooling;
c) non modificare la sequenza d'identificazione della donazione, una volta assegnata ai tessuti e alle cellule rilasciati per la circolazione, salvo che non sia necessario correggere un errore di codifica; qualsiasi rettifica va adeguatamente documentata;
d) utilizzare uno dei sistemi di codifica dei prodotti autorizzati e i relativi numeri del prodotto di tessuti e cellule inclusi nel compendio dei prodotti di tessuti e cellule dell'UE, al più tardi prima della loro distribuzione a fini di applicazioni sull'uomo;
e) utilizzare un numero specifico della sottopartita e una data di scadenza appropriati, al più tardi prima della loro distribuzione a fini di applicazioni sull'uomo; per i tessuti e le cellule per i quali non è definita, la data di scadenza è "00000000";
f) applicare il SEC sull'etichetta del prodotto in questione in modo indelebile e permanente e menzionare detto codice nei pertinenti documenti di accompagnamento, al più tardi prima della sua distribuzione ai fini di applicazioni sull'uomo; nel caso in cui le dimensioni dell'etichetta non vi permettano l'apposizione del codice unico europeo, il codice è collegato in modo univoco ai tessuti e alle cellule recanti tale etichetta tramite i documenti di accompagnamento;
g) notificare al CNT entro e non oltre 10 giorni lavorativi i casi in cui:
1) sia necessario procedere ad un aggiornamento o una correzione delle informazioni contenute nel compendio degli istituti dei tessuti dell'UE,
2) sia necessario un aggiornamento del compendio dei prodotti di tessuti e cellule dell'UE,
3) l'istituto dei tessuti rilevi una significativa inottemperanza alle prescrizioni relative al codice unico europeo per quanto riguarda i tessuti e le cellule ricevuti da altri istituti dei tessuti dell'UE;
h) predisporre apposite procedure operative in caso di apposizione non corretta del codice unico europeo sull'etichetta.
2. Il CNT provvede a:
a) assegnare il numero unico a tutti gli istituti dei tessuti autorizzati e accreditati in Italia; un istituto dei tessuti dislocato in più sedi può essere considerato un unico istituto dei tessuti in quanto si avvale di un unico sistema di assegnazione di numeri unici della donazione;
b) verificare l'assegnazione del numero della donazione, utilizzando il numero unico della donazione a livello nazionale, tramite il SIT;
c) verificare e monitorare la piena applicazione del codice unico europeo in Italia;
d) procedere alla convalida dei dati sugli istituti dei tessuti contenuti nel compendio degli istituti dei tessuti dell'UE per l'Italia e aggiornare, entro e non oltre dieci giorni lavorativi dalla scadenza del termine di cui al comma 3, il compendio medesimo, in particolare nei seguenti casi:
1) allorchè un nuovo istituto dei tessuti sia autorizzato e accreditato;
2) allorchè le informazioni sugli istituti dei tessuti siano modificate o non siano correttamente registrate nel compendio degli istituti dei tessuti dell'UE;
3) allorchè siano modificati i dati, di cui all'allegato XI-bis del presente decreto, in merito all'autorizzazione e all'accreditamento di un istituto dei tessuti, tra i quali:
3.1 l'autorizzazione e l'accreditamento per un nuovo tipo di tessuti o di cellule;
3.2 l'autorizzazione e l'accreditamento per una nuova attività prescritta;
3.3 le informazioni circa eventuali condizioni o deroghe aggiunte a una autorizzazione;
3.4 la sospensione, in tutto o in parte, di una autorizzazione o accreditamento specifici per un determinato tipo di attività o di tessuti o di cellule;
3.5 la revoca, in tutto o in parte, di una autorizzazione o accreditamento di un istituto dei tessuti;
3.6 la cessazione volontaria, in tutto o in parte, dell'attività o delle attività di un istituto dei tessuti, per le quali è stato autorizzato e accreditato;
e) informare le autorità competenti di un altro Stato membro nel caso in cui rilevi nel compendio degli istituti dei tessuti dell'UE informazioni inesatte relative all'altro Stato membro o riscontri una significativa inottemperanza alle prescrizioni relative al codice unico europeo riguardo all'altro Stato membro;
f) informare la Commissione e le altre autorità competenti nel caso in cui sia necessario a suo parere procedere ad un aggiornamento del compendio dei prodotti di tessuti e cellule dell'UE.
3. Le regioni e le province autonome sono tenute a comunicare al CNT qualsiasi modifica che incida in misura sostanziale sull'autorizzazione, sull'accreditamento degli istituti dei tessuti in questione, al fine dell'aggiornamento del compendio dell'UE. La comunicazione al CNT deve avvenire entro e non oltre dieci giorni lavorativi, decorrenti dal momento in cui avviene la modifica.
4. L'attribuzione del SEC non osta all'ulteriore applicazione di altri codici, assegnati dall'Istituto dei tessuti, finalizzati alla rintracciabilità di cellule e tessuti all'interno dell'Istituto stesso.
Art. 16. Recepimento
1. Le regioni e le province autonome di Trento e Bolzano attuano con proprio provvedimento le disposizioni di cui al presente decreto.
2. Con accordo da sancire in sede di Conferenza Stato-regioni sono stabilite le modalità e procedure necessarie a conformarsi alle prescrizioni dell'articolo 15.
Art. 16 bis. (Sospensione o revoca dell'autorizzazione e dell'accreditamento degli Istituti dei tessuti). [32]
1. La regione o la provincia autonoma competente, ai sensi dell'articolo 6, comma 5, del
Art. 17. Clausola di cedevolezza
1. In relazione a quanto disposto dall'articolo 117, quinto comma, della Costituzione e dall'articolo 16, comma 3, della
Art. 18. Disposizioni finanziarie
1. Ai nuovi o maggiori oneri di cui all'articolo 5 del presente decreto - pari a euro 1.080.000 annui a decorrere dall'anno 2010 - si provvede a valere sulle disponibilità del Fondo di rotazione di cui all'articolo 5 della
2. Il Ministro dell'economia e delle finanze è autorizzato ad apportare, con propri decreti, le occorrenti variazioni di bilancio.
3. Le attività previste dalle restanti norme del presente decreto sono svolte dalle amministrazioni competenti con le risorse umane, strumentali e finanziarie disponibili a legislazione vigente.
ALLEGATO I
CRITERI DI SELEZIONE DEL DONATORE DI TESSUTI E/O DI CELLULE
(Articolo 4, comma 1, lettera a))
I criteri di selezione dei donatori di tessuti e cellule sono basati sull'analisi dei rischi connessi all'applicazione terapeutica dei medesimi. Gli indicatori di tali rischi sono individuati mediante la raccolta dell'anamnesi clinica e comportamentale, l'esecuzione di esami fisici, test biologici, accertamento necroscopico (nel caso di donatori deceduti) e qualsiasi altro esame pertinente.
A meno che una valutazione dei rischi documentata ed approvata dalla persona responsabile dell'istituto dei tessuti lo giustifichi, un soggetto non è accettato come donatore qualora ricorra uno dei casi di seguito elencati.
1. Donatore cadavere
1.1. Criteri di esclusione
1.1.1. Causa di morte sconosciuta, (non si applica nel caso in cui l'accertamento necroscopico eseguito dopo il prelievo fornisca informazioni sulla causa di decesso che esclude i criteri di cui al presente allegato);
1.1.2. Malattie ad eziologia sconosciuta;
1.1.3. Presenza o precedenti manifestazioni di patologie maligne, eccettuato il carcinoma basocellulare primario, il carcinoma in situ della cervice uterina ed alcuni tumori primari del sistema nervoso centrale che sono valutati sulla base della documentazione scientifica. I donatori affetti da patologie maligne possono essere valutati e presi in considerazione per la donazione della cornea tranne nel caso di retinoblastoma, neoplasia ematologica e tumori maligni del segmento anteriore dell'occhio;
1.1.4. Rischio di trasmissione di malattie causate da prioni: Tale rischio riguarda, per esempio:
a) persone alle quali è stata diagnosticata la malattia di Creutzfeldt-Jakob, o la sua variante, ovvero con antecedenti familiari della malattia di Creutzfeldt-Jakob non iatrogena;
b) persone con un'anamnesi di demenza a rapida progressione, o di malattie neurologiche degenerative, comprese le patologie di origine sconosciuta;
c) riceventi ormoni derivanti dall'ipofisi umana (come gli ormoni della crescita), riceventi innesti di cornea, sclera e dura madre nonchè persone che hanno subito interventi neurochirurgici non documentati (nei quali può essere stata utilizzata la dura madre).
Per il rischio di trasmissione della variante della malattia di Creutzfeldt-Jakob non possono essere accettati donatori che sono stati sottoposti a intervento chirurgico o trasfusione di sangue o somministrazione di emoderivati nel Regno Unito dal 1980 al 1996;
1.1.5. Infezioni sistemiche che non sono state controllate al momento della donazione, comprese malattie batteriche e infezioni sistemiche virali, fungine e parassitarie o gravi infezioni locali dei tessuti e delle cellule destinati a donazioni. I donatori affetti da setticemia batterica possono essere valutati e presi in considerazione per la donazione degli occhi solo a condizione che le cornee siano destinate ad essere conservate in coltura, al fine di consentire l'individuazione di eventuali contaminazioni del tessuto;
1.1.6. Anamnesi, evidenza clinica o di laboratorio per rischio di trasmissione di HIV, epatite B acuta o cronica (tranne a persone la cui immunità è documentata), epatite C, HTLV I/II o evidenza di altri fattori di rischio connessi a tali infezioni;
1.1.7. Anamnesi positiva per malattie sistemiche autoimmuni croniche che potrebbero pregiudicare la qualità dei tessuti da prelevare;
1.1.8. Non validità dei risultati dell'esame dei campioni di sangue del donatore per:
a) emodiluizione, conformemente alle specifiche dell'allegato II, sezione 2, qualora non sia disponibile un campione prelevato prima della trasfusione;
b) trattamento a base di agenti immunosoppressivi;
1.1.9. Positività alle prove relative ad altri fattori di rischio connessi a malattie trasmissibili, sulla base di una valutazione dei rischi che tenga conto dei viaggi e dell'esposizione del donatore a tali rischi, nonchè della prevalenza locale di malattie infettive;
1.1.10. Presenza sul corpo del donatore di segni fisici che implicano rischio di malattie trasmissibili, secondo quanto descritto nell'allegato IV, punto 1.2.3;
1.1.11. Ingestione o esposizione a sostanze tossiche (quali cianuro, piombo, mercurio o oro) che possano essere trasmesse al ricevente in quantità tali da poterne compromettere la salute; 1.1.12.Vaccinazione recente con virus viventi attenuati, ove il rischio di trasmissione sia ritenuto possibile;
1.1.13. Xenotrapianti.
1.2. Criteri aggiuntivi per donatore pediatrico
1.2.1. I neonati da madri affette da HIV, o che comunque rientrano in uno dei criteri di esclusione di cui alla sezione 1.1, devono essere esclusi dalla donazione fino a che qualsiasi rischio di trasmissione dell'infezione non sia definitivamente escluso.
a) I bambini di età inferiore a 18 mesi, nati da madri affette da HIV, epatite B, epatite C o HTLV, o a rischio di contrarre l'infezione, che sono stati allattati dalle madri nei 12 mesi precedenti, non possono essere considerati donatori, indipendentemente dai risultati degli esami di laboratorio.
b) I bambini nati da madri affette da HIV, epatite B, epatite C o HTLV, o a rischio di contrarre l'infezione, che non sono stati allattati dalle madri nei 12 mesi precedenti e che non risultano affetti da HIV, epatite B, epatite C o HTLV sulla base degli esami analitici o fisici e del controllo delle cartelle cliniche, possono essere ammessi come donatori.
2. Donatore vivente
2.1. Donatore autologo
2.1.1. Nel caso di tessuti e cellule prelevati e destinati ad essere conservati o coltivati, è necessario eseguire gli stessi test di laboratorio minimi previsti per il donatore vivente allogenico. Eventuali risultati positivi dei test non comportano necessariamente il divieto di conservare, trattare e reimpiantare tessuti, cellule o qualsiasi prodotto derivato, purchè sia possibile conservarli isolatamente, al fine di evitare qualsiasi rischio di contaminazione crociata con altri innesti o di contaminazione con agenti occasionali, ovvero di miscellanea.
2.2. Donatore allogenico
2.2.1. Il donatore allogenico è selezionato sulla base dell'anamnesi clinica, delle risposte ad un questionario predisposto e del colloquio con il medico responsabile della selezione o con personale sanitario appositamente formato, operante sotto la responsabilità del predetto conformemente al punto 2.2.2. La valutazione della idoneità alla donazione, effettuata dal medico responsabile, comprende l'analisi dei fattori che possono contribuire ad individuare e ad escludere soggetti la cui donazione può costituire un rischio per la loro salute o un rischio per il ricevente, come la possibilità di trasmettere malattie. Per ogni donazione, la procedura del prelievo non deve interferire con lo stato di salute del donatore, nè comprometterlo, così come non deve interferire nè compromettere lo stato di salute della madre e del neonato il prelievo della placenta o del sangue cordonale.
2.2.2. I criteri di selezione dei donatori allogenici viventi sono documentati dall'istituto dei tessuti (o dal responsabile del trapianto in caso di distribuzione diretta al ricevente) sulla base del tipo di tessuti e cellule necessari per la donazione, delle condizioni fisiche del donatore, dell'anamnesi medica e comportamentale e dei risultati delle indagini cliniche ed esami di laboratorio relativi allo stato di salute del donatore.
2.2.3. I criteri di esclusione dalla donazione sono quelli indicati dal punto 1.1.2 al punto 1.1.13. La gravidanza e l'allattamento al seno costituiscono motivi di esclusione dalla donazione, tranne che per la placenta e il sangue cordonale. Per la donazione di cellule progenitrici ematopoietiche deve essere anche esclusa la possibilità di trasmissione di patologie ereditarie.
3. Con accordo Stato Regioni sono stabiliti i criteri di selezione dei donatori viventi e deceduti di cellule e tessuti, predisposti dal Centro Nazionale Trapianti e dal Centro Nazionale Sangue, secondo l'ambito di competenza, conformemente a quanto previsto dal presente decreto.
ALLEGATO II
ESAMI DI LABORATORIO RICHIESTI PER I DONATORI
(Articolo 5, comma 1)
1. Test di laboratorio
1.1. Il donatore di cellule e/o tessuti è sottoposto almeno ai seguenti test:
Anti-HIV-1,2
HBsAg
Anti-HBc
Anti-HCV Ab
Sifilide , come indicato al punto 1.4.
1.2. L'esame degli anticorpi HTLV-I è effettuato sui donatori che vivono in aree ad alta prevalenza del virus o ne sono originari o i cui partner sessuali provengono da tali aree, ovvero qualora i genitori del donatore siano originari di tali aree [33].
1.3. Se il test degli anticorpi anti-HBc risulta positivo e quello dell'HBsAg negativo, sono necessarie ulteriori indagini con una valutazione dei rischi per decidere l'idoneità per uso clinico. Per le cellule staminali emopoietiche midollari e periferiche e cordonali, la ricerca di anticorpi anti - HBc non è indispensabile nel caso in cui venga effettuato il test NAT per HIV, HBV e HCV, come previsto dalla normativa vigente.
1.4. Per escludere la presenza di un'infezione attiva da Treponema pallidum, deve essere applicato un algoritmo di controllo convalidato. In caso di risultato negativo al test, specifico o non specifico, i tessuti e le cellule possono essere utilizzati. Se si effettua un test non specifico, il risultato positivo non esclude il prelievo o l'utilizzo, se il test di conferma specifico sul treponema è negativo.
Per un donatore risultato positivo in un test specifico del treponema, occorre una valutazione dei rischi approfondita al fine di decidere in merito all'idoneità per uso clinico.
1.5. In determinate circostanze (quali in caso di cardiopatia reumatica, malaria, CMV, toxoplasma, EBV, Trypanosoma cruzi) possono risultare necessari ulteriori esami, in base agli antecedenti del donatore e alle caratteristiche dei tessuti o delle cellule donati .
1.6. In caso di donatori autologhi, si applica l'allegato I, punto 2.1.1.
2. Requisiti generali per la determinazione dei marcatori biologici
2.1. Gli esami sono effettuati presso laboratori autorizzati e a tal fine accreditati da parte della Regione o della Provincia autonoma, che utilizzano dispositivi diagnostici marcati CE, se richiesto. Il tipo di test impiegato deve essere convalidato per il suo scopo conformemente alle attuali conoscenze scientifiche.
2.2. I test di laboratorio sono effettuati su siero o su plasma del donatore e non su altri fluidi o secrezioni quali umore acqueo o vitreo, a meno che non sia specificamente giustificato dal punto di vista clinico, in tal caso deve essere utilizzato un test convalidato per tale fluido.
2.3. Se il potenziale donatore ha avuto emorragie e recentemente è stato sottoposto a trasfusione di sangue, suoi componenti, colloidi o cristalloidi, l'esame del sangue può non essere valido a causa dell'emodiluizione dei campioni.
E' necessario applicare un calcolo algoritmico per valutare il grado di emodiluizione nei casi seguenti:
a) campioni di sangue da donatore vivente: se il sangue, i suoi componenti e/o i colloidi sono stati iniettati entro le 48 ore precedenti il prelievo di sangue e se i cristalloidi sono stati iniettati un'ora prima del prelievo;
b) campioni di sangue da donatore cadavere: se il sangue, i suoi componenti e/o i colloidi sono stati iniettati entro le 48 ore precedenti il decesso e se i cristalloidi sono stati iniettati un'ora prima del decesso.
Gli istituti dei tessuti possono accettare tessuti e cellule di donatori con una diluizione del plasma superiore al 50 % solo se utilizzano procedure di analisi convalidate per il plasma o se dispongono di un campione precedente la trasfusione.
2.4. In caso di donatore cadavere, i campioni di sangue sono prelevati immediatamente prima del decesso, oppure, se ciò non è possibile, il prelievo dei campioni va effettuato quanto prima e in ogni caso entro 24 ore dal decesso.
2.5. a) Nel caso di donatore vivente, i campioni di sangue sono prelevati contemporaneamente alla donazione, oppure, se non è possibile, entro 7 giorni dalla donazione (si tratta del «campione della donazione»).
b) Ove tessuti e cellule di donatore allogenico vivente possano essere conservati per lunghi periodi, è necessario ripetere il prelievo dei campioni e gli esami dopo un intervallo di 180 giorni dal prelievo. In caso di ripetizione degli esami, il campione della donazione può essere prelevato sino a 30 giorni prima e 7 giorni dopo la donazione.
c) Se tessuti e cellule di donatore allogenico vivente non possono essere conservati per lunghi periodi e risulta quindi impossibile ripetere il campionamento, si applica la procedura di cui alla lettera a).
2.6. Se il «campione della donazione» di un donatore vivente, di cui alla lettera a) del punto 2.5, è anche sottoposto a test per HIV, HBV e HCV mediante la tecnica per l'amplificazione degli acidi nucleici (NAT), non è necessario ripetere l'esame dei campioni di sangue a 180 giorni dal prelievo. La ripetizione degli esami non è richiesta neppure se il trattamento comprende una fase di inattivazione convalidata per i virus interessati.
2.7. In caso di prelievo di cellule staminali del midollo osseo e del sangue periferico, ivi compreso il sangue da cordone ombelicale, i campioni di sangue sono prelevati a fini di analisi nei 30 giorni precedenti la donazione e sono testati secondo le disposizioni vigenti in materia di attività trasfusionali.
2.8 Nel caso di donatori neonati, i test biologici possono essere effettuati sulla madre del donatore, al fine di evitare interventi medici inutili sul bambino.
ALLEGATO III
CRITERI DI SELEZIONE ED ESAMI DI LABORATORIO RICHIESTI
PER I DONATORI DI CELLULE RIPRODUTTIVE
(Articolo 4, lettera b) - Articolo 5, comma 2)
1. Donazione del partner destinata all'impiego diretto
In caso di donazione da parte del partner di cellule riproduttive destinate all'impiego diretto, non occorre applicare i criteri di selezione dei donatori nè effettuare gli esami di laboratorio, salvo che non vi sia rischio di trasmissione di infezioni a terzi.
2. Donazione del partner (casi diversi dall'impiego diretto) e donazione da persone diverse dal partner [34]
A. Donazione del partner (casi diversi dall'impiego diretto)
Le cellule riproduttive lavorate e/o conservate e le cellule riproduttive che daranno origine ad embrioni sono conformi ai seguenti criteri.
2.1. Il medico clinico che segue il donatore deve definire e documentare, sulla base dell'anamnesi dello stesso e delle indicazioni terapeutiche, una giustificazione della donazione e la sua sicurezza per il ricevente e per gli eventuali bambini che possono nascere.
2.2. Al fine di valutare il rischio di contaminazioni incrociate, vanno effettuati i seguenti test biologici:
Anti-HIV-1,2, HBsAg, Anti-HBc, Anti-HCV Ab.
Nel caso di sperma lavorato per l'inseminazione intrauterina non destinato alla conservazione, e a condizione che l'istituto dei tessuti possa dimostrare che il rischio di contaminazione incrociata e di esposizione del personale sia stato scongiurato tramite il ricorso a procedure convalidate, è possibile rinunciare all'obbligatorietà dello svolgimento di test biologici.
2.3. Ove i risultati dei test dell'HIV 1 e 2, dell'epatite B o dell'epatite C siano positivi oppure non disponibili, o qualora risulti che il donatore comporta un rischio d'infezione, occorre predisporre un sistema di conservazione separata.
2.4. L'esame degli anticorpi HTLV-I va effettuato sui donatori che vivono in aree ad alta prevalenza o ne sono originari o i cui partner sessuali provengono da tali aree, ovvero qualora i genitori del donatore siano originari di tali aree.
2.5. In determinate circostanze, possono risultare necessari ulteriori esami, in base ai viaggi e all'esposizione del donatore a fattori di rischio e alle caratteristiche dei tessuti o delle cellule donati (per es. in caso di cardiopatia reumatica, malaria, CMV, T. cruzi).
2.6. I risultati positivi non impediscono necessariamente la donazione del partner in base alla normativa vigente.
B. Donazione da persone diverse dal partner.
La donazione di cellule riproduttive da parte di persone diverse dal partner deve soddisfare i seguenti criteri e modalità.
2.1. La donazione di cellule riproduttive è consentita ai soggetti di sesso maschile di età non inferiore ai diciotto anni e non superiore ai quaranta anni e ai soggetti di sesso femminile di età non inferiore ai venti anni e non superiore ai trentacinque anni. Le cellule riproduttive donate da un medesimo donatore non possono determinare più di dieci nascite. Tale limite può essere derogato esclusivamente nei casi in cui una coppia, che abbia già avuto un figlio tramite procreazione medicalmente assistita di tipo eterologo, intenda sottoporsi nuovamente a tale pratica utilizzando le cellule riproduttive del medesimo donatore.
La selezione dei donatori avviene sulla base dell'anamnesi sanitaria e medica compiuta anche sulla base di un questionario cui gli stessi sono sottoposti e di un colloquio individuale con il medico responsabile della selezione o con personale sanitario appositamente formato, anche in materia di protezione dei dati personali, operante sotto la responsabilità del predetto medico responsabile. Tale valutazione deve comprendere fattori rilevanti che possono contribuire a individuare e ad escludere le persone la cui donazione può costituire un rischio sanitario per gli altri, come la possibilità di trasmettere malattie, rischi sanitari per i donatori stessi, quali ad esempio superovulazione, possibili reazioni alla somministrazione di sedativi o rischi associati all'intervento per il prelievo di ovociti, oppure conseguenze psicologiche per il donatore.
La donatrice di ovociti non può essere sottoposta ad un numero di cicli di stimolazioni ovariche superiore a sei. In ogni caso, a tutela della salute della donatrice devono essere previsti specifici monitoraggi periodici.
I limiti relativi all'età dei donatori, al numero delle donazioni degli ovociti e dei gameti maschili e al numero delle stimolazioni ormonali cui può essere sottoposta la donatrice, nonchè al numero delle nascite scaturenti dal medesimo donatore, sono oggetto di verifica almeno triennale sulla base dei risultati dell'esperienza, della ricerca e delle migliori pratiche della scienza medica seguite anche in sede internazionale, avvalendosi, per la verifica dei limiti al numero delle nascite scaturenti dal medesimo donatore, anche delle competenze dell'Istituto nazionale di statistica (ISTAT), ai fini dei successivi aggiornamenti del presente regolamento.
Il trattamento dei dati personali è effettuato in conformità ai principi di finalità del trattamento, di indispensabilità e necessità, di proporzionalità, pertinenza e non eccedenza dei dati personali trattati e nel rispetto di quanto previsto dal
2.2. I donatori di cellule riproduttive devono risultare negativi ai test per l'HIV 1 e 2, l'HCV, l'HBV e la sifilide, effettuati su un campione di siero o di plasma conformemente all'allegato II, punto 1.1. I donatori di sperma devono inoltre risultare negativi al test per la clamidia, effettuato su un campione di urina mediante la tecnica per l'amplificazione degli acidi nucleici (NAT).
2.3. L'esame degli anticorpi HTLV-I va effettuato sui donatori che vivono in aree ad alta prevalenza o ne sono originari o i cui partner sessuali provengono da tali aree, ovvero qualora i genitori dei medesimi siano originari di tali aree.
2.4. In determinate circostanze, sulla base delle valutazioni del medico, possono risultare necessari ulteriori esami in base agli antecedenti del donatore e alle caratteristiche dei tessuti o delle cellule donati.
2.5. In caso di donatori autologhi, si applicano le norme di cui all'allegato I, punto 2.1.1.
2.6. Ai fini dello screening genetico di geni autosomici recessivi risultati prevalenti nel contesto etnico del donatore in base a evidenze scientifiche internazionali, nonchè di una valutazione del rischio di trasmissione di patologie ereditarie che risultano presenti nella famiglia del donatore, sono effettuati una visita di genetica medica con relazione scritta, il test per la fibrosi cistica ed eventuali ulteriori esami, compreso l'esame del cariotipo, ritenuti necessari sulla base della predetta visita, previa acquisizione dell'autorizzazione da parte del medesimo donatore, nel rispetto della normativa vigente sul consenso informato e delle disposizioni europee e nazionali in materia di trattamento dei dati personali, che consentono il trattamento dei dati relativi alla salute e di quelli genetici, in presenza di una delle condizioni di cui al paragrafo 2 dell'articolo 9 del
2.7. Alla coppia che accede alle tecniche di procreazione assistita di tipo eterologo vanno fornite informazioni dettagliate e illustrati con chiarezza i rischi associati ad essa, nonchè le misure adottate per attenuarli. In particolare, la coppia deve essere informata in merito agli esami clinici cui è stato sottoposto il donatore, dei relativi test effettuati e del fatto che tali esami non possono garantire, in modo incontrovertibile, l'assenza di patologie per il nascituro. Nel rispetto delle disposizioni vigenti in materia di trattamento dei dati personali, è salvaguardata la riservatezza del donatore, specie mediante l'adozione di un sistema di identificazione indiretta del medesimo.
3. Prescrizioni generali da osservare per la determinazione dei marcatori biologici [35]
3.1. I test di cui al punto 2 vanno effettuati conformemente all'allegato II, punti 2.1 e 2.2.
3.2. Nel caso delle donazioni del partner (casi diversi dall'impiego diretto), i campioni di sangue vanno prelevati non oltre novanta giorni prima del prelievo ovvero della raccolta dei gameti e ripetuti ogni sei mesi durante il trattamento. Nel caso di crioconservazione dei gameti e degli embrioni non è necessaria la ripetizione dei test.
3.3. Nel caso delle donazioni di persone diverse dal partner, i campioni di sangue vanno prelevati al momento della donazione.
Nel caso della donazione di gameti femminili, il momento della donazione può essere considerato il primo giorno dell'inizio della stimolazione e i campioni di sangue possono essere raccolti in questo momento.
3.4. I gameti donati da persone diverse dal partner sono messi in quarantena per almeno centottanta giorni e successivamente occorre ripetere gli esami. Non si ricorre alla quarantena se il campione di sangue prelevato al momento della donazione viene sottoposto anche a test con tecnica di amplificazione nucleica (NAT) per HIV, HBV, e HCV, ferma restando l'effettuazione dei test seriologici al momento della donazione. La ripetizione degli esami non è richiesta neppure se il trattamento comprende una fase di inattivazione convalidata per i virus interessati.
In ogni caso, i risultati dei test sui donatori devono essere disponibili prima dell'utilizzo dei gameti.
ALLEGATO IV
PROCEDURE RELATIVE ALLA DONAZIONE E ALL'APPROVVIGIONAMENTO DI TESSUTI E/O DI CELLULE E RICEVIMENTO PRESSO L'ISTITUTO DEI TESSUTI
(Articolo 6)
1. Procedure relative alla donazione e all'approvvigionamento
1.1. Consenso alla donazione e identificazione del donatore
1.1.1. Prima di procedere all'approvvigionamento di tessuti e cellule, un sanitario qualificato ed autorizzato a tal fine conferma ed indica:
a) che la manifestazione di volontà al prelievo è stata ottenuta in conformità a quanto previsto dall'articolo 13 del
b) le modalità attraverso le quali è stata accertata l'identità del donatore e da chi.
1.1.2. Nel caso di donatore vivente, il medico responsabile della selezione, o personale sanitario appositamente formato operante sotto la responsabilità del predetto, che raccoglie informazioni sull'anamnesi, si accerta che il donatore nel corso del colloquio:
a) abbia compreso le informazioni fornite;
b) abbia avuto l'opportunità di porre domande e abbia ricevuto risposte esaurienti;
c) abbia confermato che tutte le informazioni e le risposte fornite sono veritiere.
1.2. Valutazione del donatore (questa sezione non si applica alle donazioni di cellule riproduttive da parte del partner e ai donatori autologhi)
1.2.1. Il medico responsabile della selezione, o personale sanitario appositamente formato operante sotto la responsabilità del predetto, raccoglie e registra tutte le informazioni relative all'anamnesi medica e comportamentale del donatore secondo le disposizioni di cui alla sezione 1.4.
1.2.2. Informazioni esaustive sullo stato di salute del donatore possono essere ottenute attraverso diverse fonti, compreso almeno un colloquio diretto con il donatore (nel caso di donatore vivente), e se applicabile, anche attraverso:
a) la cartella clinica del donatore,
b) un colloquio con persona che conosceva bene il donatore (nel caso di donatore deceduto)
c) un colloquio con il medico curante,
d) un colloquio con il medico generico,
e) il referto dell'accertamento necroscopico.
1.2.3. In caso di donatore cadavere e, ove risulti giustificato, nel caso di donatore vivente, deve essere eseguito un esame fisico del corpo, al fine di rilevare eventuali segni sufficienti di per sè ad escludere la donazione o comunque da valutare alla luce della storia clinica e comportamentale del medesimo.
1.2.4. I dati completi relativi al donatore sono esaminati, valutati e firmati dal medico responsabile della selezione ai fini del giudizio di idoneità.
1.3. Procedure relative all'approvvigionamento di tessuti e cellule
1.3.1. Le procedure di approvvigionamento debbono essere adeguate al tipo di donatore e al tipo di tessuti o cellule donati. Deve, in ogni caso, essere garantita la sicurezza del donatore vivente.
1.3.2. Le procedure di approvvigionamento si svolgono in modo tale da salvaguardare le proprietà dei tessuti e delle cellule necessarie per l'uso clinico finale e nel contempo da ridurre i rischi di contaminazione microbiologica durante il processo, in particolare quando tessuti e cellule non possono essere sterilizzati.
1.3.3. In caso di donazioni da donatore cadavere, l'accesso all'area prelievo deve essere limitato. E' necessario disporre di un campo sterile locale, dotato di teli sterili. L'abbigliamento del personale autorizzato al prelievo deve essere adeguato al tipo di prelievo, deve essere fornito di abiti e guanti sterili, di schermi per il viso o di maschere di protezione.
1.3.4. In caso di donatore cadavere, è necessario indicare il luogo dell'approvvigionamento e l'intervallo di tempo intercorso tra il decesso e il prelievo, al fine di garantire che siano salvaguardate le proprietà biologiche e fisiche necessarie dei tessuti o delle cellule.
1.3.5. Dopo il prelievo dei tessuti o delle cellule, il corpo del donatore cadavere deve essere ricomposto in modo che sia ripristinato il più possibile l'aspetto anatomico originario.
1.3.6. Qualsiasi incidente avvenuto durante il prelievo che abbia danneggiato o che possa aver danneggiato il donatore vivente nonchè il risultato delle indagini volte ad accertarne le cause sono registrati ed analizzati.
1.3.7. E' necessario predisporre adeguate misure e procedure tese ad evitare il rischio di contaminazione dei tessuti o delle cellule da parte di personale eventualmente affetto da malattie trasmissibili.
1.3.8. Per il prelievo di tessuti e cellule sono utilizzati strumenti e dispositivi sterili. Tali strumenti e dispositivi devono essere di qualità, convalidati o espressamente certificati ed a tal fine abitualmente utilizzati.
1.3.9. Nel caso di impiego di strumenti riutilizzabili, deve essere predisposta una procedura convalidata per la pulizia e sterilizzazione, al fine di eliminare eventuali agenti infettivi. 1.3.10. Ove possibile, sono impiegati soltanto dispositivi medici marcati CE, il personale sanitario addetto alle attività di prelievo riceve adeguata formazione sull'utilizzo di tali dispositivi.
1.4. Documentazione del donatore
1.4.1. Per ogni donatore deve essere predisposta una cartella contenente:
a) dati anagrafici (nome, cognome, luogo e data di nascita). Se nella donazione sono coinvolti una madre e un bambino, i dati relativi alla madre e al bambino.
b) età, sesso, anamnesi clinica e comportamentale (le informazioni raccolte devono essere sufficienti a consentire l'applicazione dei criteri di esclusione se necessario);
c) se necessario, l'esito dell'esame fisico del corpo;
d) formula relativa all'emodiluizione, se richiesta;
e) modulo relativo al consenso o alla manifestazione di volontà alla donazione;
f) dati clinici, risultati di esami di laboratorio e risultati di altri test effettuati;
g) nel caso in cui sia stato eseguito accertamento necroscopico, i risultati devono essere annotati nella cartella (nel caso di tessuti e cellule che non possono essere conservati per lunghi periodi, deve essere registrato un preliminare resoconto orale dell'accertamento e annotato nella documentazione che l'autopsia è in corso);
h) per i donatori di cellule progenitrici ematopoietiche, va documentata l'idoneità del donatore al ricevente scelto.
Per donazioni senza un preciso destinatario, ove l'organizzazione responsabile dell'approvvigionamento abbia un accesso limitato ai dati del ricevente, al centro trapianti devono essere forniti i dati del donatore necessari a confermare l'idoneità.
1.4.2. L'organizzazione in cui si effettua il prelievo trasmette all'istituto dei tessuti relazione e documentazione inerenti al prelievo. Tale documentazione deve comprendere almeno:
a) denominazione e indirizzo dell'istituto dei tessuti cui sono destinati i tessuti o le cellule;
b) dati identificativi del donatore, nonchè il modo in cui è stato identificato e da chi;
c) descrizione e identificazione dei tessuti e delle cellule prelevati (compresi i campioni destinati alle analisi);
d) generalità del sanitario responsabile del prelievo, compresa la firma;
e) data, ora (se necessario, d'inizio e di conclusione) e luogo del prelievo nonchè procedura impiegata (POS) ed eventuali incidenti verificatisi; se necessario descrizione dell'area fisica in cui è stato effettuato il prelievo;
f) nel caso di donatore cadavere, condizioni in cui viene conservato lo stesso: refrigerato (o no), se sì: ora d'inizio e fine della refrigerazione, nonchè data e ora della morte;
g) numero del lotto o d'identificazione dei reagenti nonchè soluzioni adottate durante il trasporto.
Qualora lo sperma sia prelevato a casa, la relazione sul prelievo deve indicarlo e si deve figurare solo:
a) denominazione e indirizzo dell'istituto dei tessuti cui sono destinati i tessuti o le cellule;
b) dati d'identificazione del donatore.
La data e l'ora del prelievo possono essere indicati, ove possibile.
1.4.3. Tutti i registri devono essere chiari e leggibili, protetti da modifiche non autorizzate, conservati e facilmente recuperabili nella forma originale durante tutto il periodo di conservazione, conformemente alle disposizioni vigenti in materia di tutela della riservatezza.
1.4.4. I registri dei donatori, necessari ai fini di una completa tracciabilità, sono conservati per almeno 30 anni dopo l'uso clinico o dopo la scadenza o eliminazione del tessuto o cellula in un archivio adeguato, approvato dall'autorità regionale competente.
1.5. Confezionamento
1.5.1. Dopo l'approvvigionamento, tutti i tessuti e le cellule prelevati sono confezionati in modo da evitare il rischio di contaminazione e conservati a temperature che garantiscono il mantenimento delle loro caratteristiche e funzioni biologiche. Il confezionamento deve inoltre evitare la contaminazione del personale incaricato della sua effettuazione nonchè di quello incaricato del trasporto di tessuti e cellule.
1.5.2. Le cellule o i tessuti confezionati sono spediti in contenitore idoneo al trasporto di materiali biologici e in grado di salvaguardare la sicurezza e la qualità dei tessuti e delle cellule in esso contenuti.
1.5.3. Eventuali campioni di tessuti o di sangue che accompagnano i materiali a fini di analisi devono essere accuratamente etichettati per garantire l'identificazione del donatore e devono recare l'indicazione dell'ora in cui sono stati prelevati.
1.6. Etichettatura dei tessuti o delle cellule prelevati
Al momento del prelievo, ogni imballaggio contenente tessuti e cellule deve essere etichettato. Il contenitore primario dei tessuti o delle cellule deve recare l'identificazione o il codice della donazione e l'indicazione del tipo di tessuti o di cellule. Ove le dimensioni del contenitore lo consentano, sono inoltre fornite le seguenti informazioni:
a) data (e, ove possibile, ora) della donazione;
b) avvertenze;
c) tipo di additivi (se pertinente);
d) in caso di donatori autologhi, l'etichetta deve recare la dicitura «solo per uso autologo»;
e) in caso di donazioni con destinatario, l'etichetta deve identificare il ricevente scelto. Se le informazioni di cui alle precedenti lettere da a) a e) non possono essere indicate nell'etichetta del contenitore primario, vanno fornite in un foglio separato che accompagna il contenitore.
1.7. Etichettatura del contenitore usato per il trasporto
Ove i tessuti e le cellule siano trasportati da un intermediario, ogni contenitore usato per il trasporto deve essere etichettato e recare le seguenti indicazioni:
a) le diciture: TESSUTI E CELLULE e MANIPOLARE CON CAUTELA;
b) l'identificazione dell'istituto dal quale viene spedito l'imballaggio (indirizzo e numero di telefono) e persona da contattare in caso di problemi;
c) l'identificazione dell'istituto dei tessuti di destinazione (indirizzo e numero di telefono) e persona da contattare per la consegna del contenitore;
d) data e ora d'inizio del trasporto;
e) descrizione delle condizioni di trasporto con riguardo alla qualità e alla sicurezza dei tessuti e delle cellule;
f) per tutti i prodotti cellulari, occorre aggiungere la seguente dicitura: NON IRRADIARE;
g) ove un prodotto risulti positivo a un marcatore di una malattia infettiva, la seguente dicitura: RISCHIO BIOLOGICO;
h) in caso di donatori autologhi, la seguente dicitura: SOLO PER USO AUTOLOGO;
i) avvertenze sulle condizioni di conservazione (come NON CONGELARE).
2. Ricevimento dei tessuti e delle cellule presso l'istituto dei tessuti
2.1. Quando i tessuti e le cellule prelevati arrivano presso l'istituto dei tessuti, occorre effettuare una verifica documentata della conformità dei materiali inviati, comprese le condizioni di trasporto, l'imballaggio, l'etichettatura nonchè la documentazione e i campioni acclusi, alle disposizioni del presente allegato e alle specifiche dell'istituto ricevente.
2.2. Ciascun istituto deve assicurare che i tessuti e le cellule ricevuti siano tenuti in quarantena finchè tali materiali e la relativa documentazione siano stati ispezionati o altrimenti verificati secondo le prescrizioni. L'esame delle informazioni inerenti al donatore e al prelievo nonchè la conseguente accettazione della donazione sono effettuati da personale debitamente autorizzato.
2.3. Ciascun istituto dei tessuti deve disporre di linee di condotta e di specifiche documentate in base alle quali verifica ogni invio di tessuti e di cellule, inclusi i campioni. Tale documentazione comprende le prescrizioni tecniche citate e altri criteri che l'istituto dei tessuti ritiene essenziale ai fini della salvaguardia di un'adeguata qualità.
L'istituto dei tessuti deve disporre di procedure documentate per la gestione e la separazione dei materiali non conformi o con risultati delle analisi incompleti, al fine di garantire che non sussistono rischi di contaminazione per altri tessuti e cellule lavorati, conservati o stoccati.
2.4. Tra i dati che l'istituto dei tessuti deve registrare (tranne in caso di donatori di cellule riproduttive destinate alla donazione al partner) rientrano:
a) l'assenso o autorizzazione, in particolare lo scopo per cui possono essere impiegati i tessuti e le cellule (ovvero uso terapeutico o uso di sperimentazione clinica, oppure uso sia terapeutico che di sperimentazione clinica) e qualsiasi istruzione specifica relativa all'eliminazione se i tessuti o le cellule non sono utilizzati per scopo a cui erano destinati;
b) tutta le documentazione prescritta riferita all'approvvigionamento e alla selezione del donatore, secondo quanto indicato nella sezione sulla documentazione del donatore;
c) i risultati dell'esame fisico, dei test di laboratorio e degli altri esami (quali il referto dell'autopsia, ove sia stata effettuata ai sensi del punto 1.2.2.);
d) per i donatori allogenici, un riesame debitamente documentato dell'intera valutazione del donatore sulla base dei criteri di selezione effettuato da personale autorizzato ed esperto;
e) in caso di colture di cellule destinate all'uso autologo, indicazione di eventuali allergie a medicinali del ricevente (per esempio agli antibiotici).
2.5. Per quanto riguarda le cellule riproduttive destinate alla donazione al partner, i dati che l'istituto dei tessuti deve registrare comprendono:
a) l'autorizzazione, in particolare lo scopo per cui possono essere impiegati i tessuti e le cellule (per esempio, solo per uso terapeutico o per sperimentazione clinica) e qualsiasi istruzione specifica relativa all'eliminazione, se i tessuti o le cellule non sono utilizzati per scopo a cui erano destinati;
b) generalità e caratteristiche del donatore: tipo di donatore, età, sesso, presenza di fattori di rischio [36];
c) luogo del prelievo;
d) tessuti e cellule prelevati e relative caratteristiche.
ALLEGATO V
Prescrizioni per l'autorizzazione e l'accreditamento degli istituti dei tessuti
(Articolo 8)
A. Organizzazione e gestione
1. L'ente a cui afferisce l'Istituto dei tessuti ne designa la persona responsabile, che abbia le qualifiche e le responsabilità di cui all'articolo 17 del
2. L'istituto dei tessuti si dota di una struttura organizzativa e di procedure operative adeguati alle attività per le quali si chiede l'autorizzazione e l'accreditamento; un organigramma definisce chiaramente i rapporti in materia di responsabilità e di obblighi di riferire.
3. Ogni istituto dei tessuti, conformemente a quanto previsto dall'art. 17, c. 1, lett. a), del suddetto
4. Un sistema documentato di gestione della qualità è applicato alle attività per le quali si richiede l'autorizzazione e l'accreditamento, conformemente a quanto previsto dal presente decreto.
5. L' istituto dei tessuti garantisce l'individuazione e la minimizzazione dei rischi, ivi compresi quelli specificamente relativi alle procedure, ambiente e stato di salute del personale dell'istituto dei tessuti, inerenti all'uso e alla manipolazione di materiale biologico, coerentemente con il mantenimento di qualità e sicurezza adeguate alla destinazione prevista dei tessuti e cellule.
6. Gli accordi tra istituti dei tessuti e terzi sono stipulati in conformità a quanto previsto dall'articolo 24 del
7. Con la supervisione della persona responsabile, è predisposto un sistema documentato per confermare la conformità di tessuti e cellule ad adeguate specifiche di sicurezza e qualità per il rilascio e la distribuzione.
8. In caso di cessazione delle attività gli accordi conclusi e le procedure adottate in conformità ai disposti dell'articolo 21, comma 5, del
9. Presso ogni istituto dei tessuti è predisposto un sistema documentato che garantisce l'identificazione di ciascuna unità di tessuto o cellule in tutte le fasi delle attività per le quali si chiede l'autorizzazione e l'accreditamento.
B. Personale
1. L' istituto dei tessuti è dotato di personale in numero sufficiente e qualificato per lo svolgimento dei compiti assegnati. La competenza del personale è valutata ad intervalli di tempo adeguati, specificati nell'ambito del sistema di qualità.
2. Le funzioni di tutto il personale sono regolate in modo da risultare chiare, documentate ed aggiornate. I relativi compiti, competenze e responsabilità sono definiti in modo da risultare ben documentati e compresi.
3. L'istituto dei tessuti garantisce una formazione iniziale del personale e gli aggiornamenti occorrenti nel caso di modifica delle procedure o di sviluppo delle conoscenze scientifiche, nonchè possibilità di adeguata crescita professionale.
Il programma di formazione garantisce e documenta che ciascun soggetto:
a) abbia dimostrato competenza nello svolgimento dei compiti assegnati;
b) abbia conoscenza e comprensione adeguate dei procedimenti e dei principi scientifici e tecnici afferenti ai compiti assegnati;
c) comprenda il quadro organizzativo, il sistema di qualità e le norme di salute e sicurezza dell'istituto in cui opera;
d) sia adeguatamente informato del più ampio contesto etico, legislativo e normativo del proprio lavoro.
C. Attrezzature e materiali
1. La progettazione e la manutenzione delle attrezzature e i materiali corrispondono alle destinazioni d'uso previste e sono predisposte in modo da minimizzare ogni rischio per i riceventi e il personale.
2. Tutte le attrezzature e i dispositivi tecnici critici sono identificati e convalidati, periodicamente ispezionati e preventivamente sottoposti a manutenzione conformemente alle istruzioni del fabbricante. Le attrezzature o i materiali che incidono su parametri critici di lavorazione o stoccaggio (ad esempio temperatura, pressione, numero di particelle, livello di contaminazione microbica) sono identificati e sottoposti in modo adeguato a osservazioni, vigilanza, allarmi ed agli eventuali interventi correttivi necessari per individuare le disfunzioni e i difetti e per garantire che i parametri critici rimangano costantemente al di sotto dei limiti accettabili. Tutte le attrezzature che dispongono di una funzione di misurazione critica sono tarate su un parametro di riferimento reperibile, qualora esista [37].
3. Le attrezzature nuove e quelle riparate sono controllate al momento dell'installazione e collaudate prima dell'uso. I risultati dei controlli sono documentati.
4. Periodicamente è necessario procedere alla manutenzione, alla pulizia, alla disinfezione e all'igienizzazione di tutte le attrezzature critiche e alla registrazione delle operazioni effettuate.
5. Per ogni attrezzatura critica è necessario disporre di norme di funzionamento, con indicazioni dettagliate di come intervenire in caso di disfunzioni o guasti.
6. Le norme per le attività di cui si chiede l'autorizzazione e l'accreditamento indicano dettagliatamente le specifiche di tutti i materiali e i reagenti critici. Sono in particolare definite le specifiche per gli additivi (ad esempio soluzioni) e i materiali d'imballaggio. I reagenti e i materiali critici corrispondono alle prescrizioni e alle specifiche documentate e, se del caso, alle prescrizioni del
D. Servizi e locali
1. Un istituto dei tessuti possiede servizi adeguati allo svolgimento delle attività per le quali è richiesta l'autorizzazione e l'accreditamento, in conformità ai parametri stabiliti dal presente decreto .
2. Quando tali attività comprendono la lavorazione di tessuti e cellule a contatto con l'ambiente, le stesse si svolgono in un ambiente di specifica qualità e pulizia dell'aria, al fine di minimizzare i rischi di contaminazione, compresa la contaminazione incrociata tra donazioni. L'efficacia delle misure intraprese è convalidata e controllata.
3. Fatte salve le disposizioni indicate al punto 4, se i tessuti o le cellule vengono a contatto con l'ambiente durante la lavorazione senza essere poi sottoposti a procedimento di inattivazione microbica, per la qualità dell'aria è prescritto quale requisito un numero di particelle e un numero di colonie microbiche equivalente a quelli di grado A di cui all'allegato 1 della guida europea alle buone pratiche di fabbricazione (Good Manufacturing Practice: GMP), e al
4. Condizioni ambientali meno rigorose di quelle sopraindicate possono essere accettabili qualora:
a) si applichi un procedimento convalidato di inattivazione microbica o di sterilizzazione finale, oppure
b) sia dimostrato che il contatto con un ambiente di grado A ha effetti nocivi sulle proprietà richieste per i tessuti o cellule di cui si tratta, oppure
c) sia dimostrato che le modalità e il percorso di applicazione di tessuti o cellule al ricevente comportano un rischio di trasmettere al ricevente infezioni batteriche o fungine, notevolmente inferiore rispetto al trapianto di cellule e tessuti, oppure
d) non sia tecnicamente possibile eseguire il procedimento richiesto in un ambiente di grado A (ad esempio perchè nella zona di lavorazione occorrono attrezzature specifiche non del tutto compatibili con il grado A).
5. Ai fini di quanto prescritto nelle lettere a), b), c) e d) del punto 4 riguardo all'ambiente, è necessario dimostrare e documentare che l'ambiente prescelto corrisponda alla qualità e sicurezza richieste, prendendo almeno in considerazione la destinazione prevista, le modalità di applicazione e lo stato immunitario del ricevente.
In ciascun reparto dell'istituto dei tessuti sono resi disponibili indumenti ed attrezzature adeguati per la protezione e l'igiene personali, insieme ad istruzioni scritte relative all'igiene e all'abbigliamento.
6. Quando le attività per le quali si chiede l'autorizzazione e l'accreditamento comportano lo stoccaggio di tessuti e cellule, sono definite le condizioni di stoccaggio necessarie per mantenere le proprietà richieste per i tessuti e cellule, compresi i corrispondenti parametri, relativi a temperatura, umidità o qualità dell'aria.
7. I parametri critici, quali: temperatura, umidità, qualità dell'aria, sono essere controllati, sorvegliati e registrati per dimostrarne la corrispondenza con le specifiche condizioni di stoccaggio.
8. E' necessario predisporre servizi di stoccaggio che separino e distinguano nettamente i tessuti e le cellule precedenti il rilascio/in quarantena da quelli rilasciati e da quelli scartati, al fine di prevenirne la confusione e la contaminazione incrociata. Nei locali di stoccaggio di tessuti e cellule sia in quarantena che rilasciati, è necessario predisporre zone o dispositivi di stoccaggio fisicamente separati o isolamenti di sicurezza all'interno del dispositivo per la tenuta di determinati tessuti e cellule prelevati conformemente a criteri speciali.
9. L'istituto dei tessuti dispone di un regolamento e di procedure scritte per l'accesso controllato, la pulizia, la manutenzione e lo smaltimento dei rifiuti, nonchè per garantire la riorganizzazione della prestazione dei servizi in situazioni di emergenza.
E. Documentazione e registrazioni
1. L'istituto dei tessuti organizza un sistema in grado di fornire la documentazione, chiaramente definita ed efficace, registrazioni, schede corrette nonchè procedure operative standard (POS) per le attività di cui si chiede l'autorizzazione e l'accreditamento. Tutti i documenti sono periodicamente verificati ed essere conformi ai parametri indicati dal presente decreto. Il sistema garantisce la standardizzazione dell'attività svolta e la rintracciabilità di tutte le sue fasi, ovvero codifica, idoneità di donatori, approvvigionamento, lavorazione, conservazione, stoccaggio, trasporto, distribuzione o smaltimento, compresi gli aspetti relativi al controllo di qualità e alla garanzia di qualità.
2. Il materiale, le attrezzature e il personale coinvolti in ogni attività critica sono identificati e registrati.
3. Negli istituti dei tessuti tutte le modifiche dei documenti sono verificate, datate, approvate, documentate ed eseguite da personale a tal fine autorizzato.
4. E' istituita una procedura scritta per il controllo dei documenti, in grado di fornire la storia delle loro verifiche e modifiche, garantendo nel contempo l'utilizzo solo della versione in corso.
5. E' necessario dimostrare l'attendibilità delle registrazioni e che le stesse rappresentano correttamente i risultati.
6. Le registrazioni sono rese leggibili, indelebili e possono essere manoscritte, con la possibilità di avvalersi di altro sistema convalidato, quale il supporto elettronico o il microfilm.
7. Fatto salvo quanto disposto dall'articolo 14 (9), comma 2, tutte le registrazioni, dati grezzi compresi, critiche per la sicurezza e la qualità dei tessuti e cellule sono conservate, per garantirne l'accessibilità, per almeno 10 anni dopo la data di scadenza, l'uso clinico o lo smaltimento.
8. Le registrazioni soggiacciono alle prescrizioni dettate dall'articolo 14 del
F. Verifica della qualità
1. L'istituto dei tessuti predispone un sistema di verifica delle attività per le quali si richiede l'autorizzazione e l'accreditamento. Le verifiche, finalizzate ad accertare l'osservanza dei protocolli approvati e delle prescrizioni normative, sono eseguite in modo autonomo almeno ogni due anni da persone espressamente qualificate e competenti. I risultati e gli interventi correttivi sono documentati.
2. Gli scostamenti rispetto ai parametri di qualità e sicurezza richiesti sono oggetto di indagini documentate, comprendenti anche decisioni relative ad eventuali interventi correttivi e preventivi. Il destino dei tessuti e delle cellule non conformi è deciso seguendo procedure scritte con la supervisione della persona responsabile, e viene poi registrato. Tutti i tessuti e le cellule interessati sono identificati, in modo da poterne rispondere.
3. Gli interventi correttivi sono documentati, avviati e completati con puntualità ed efficacia. L'efficacia degli interventi preventivi e correttivi intrapresi è oggetto di specifica valutazione.
4. L'istituto dei tessuti predispone procedimenti di verifica del funzionamento del sistema di gestione della qualità per garantirne il progresso costante e sistematico.
ALLEGATO VI
Prescrizioni per l'autorizzazione di procedimenti di preparazione di tessuti e cellule negli istituti di tessuti
(Articolo 9 )
1. L'autorità regionale competente autorizza ogni procedimento di preparazione di tessuti e cellule dopo aver valutato i criteri di selezione del donatore e le procedure di approvvigionamento, i protocolli relativi a ciascuna fase del procedimento, i criteri di gestione della qualità e i criteri quantitativi e qualitativi definitivi per i tessuti e le cellule. Tale valutazione si basa almeno sulle prescrizioni del presente allegato.
A. Ricevimento dei tessuti e cellule
L'istituto dei tessuti riceve tessuti e cellule prelevati rispondenti ai requisiti previsti dal presente decreto .
B. Lavorazione
Nel caso in cui le attività per le quali si chiede l'autorizzazione e l'accreditamento comprendano la lavorazione di tessuti e cellule, l'istituto dei tessuti si attiene alle procedure e ai criteri seguenti:
1. Le procedure di lavorazione critiche sono convalidate e tali da non rendere i tessuti o le cellule clinicamente inefficaci o nocivi per il ricevente. La convalida si basa su studi eseguiti dall'istituto stesso o su dati tratti da studi pubblicati, o, nel caso di procedure di lavorazione pienamente affermate, sulla valutazione retrospettiva dei risultati clinici relativi all'impiego di tessuti forniti dall'istituto.
2. E' prescritta la dimostrazione che il procedimento di convalida possa essere svolto in modo coerente ed efficace nell'ambito dell'istituto dei tessuti ad opera del personale ivi operante.
3. Le procedure sono documentate nelle POS, che si attengono al metodo convalidato e ai parametri stabiliti dal presente decreto, conformemente all'allegato V, parte E, punti da 1 a 4.
4.L'istituto dei tessuti garantisce che tutti i procedimenti si svolgano in conformità alle POS approvate.
5. Qualora ai tessuti o cellule venga applicato un procedimento di inattivazione microbica, esso va specificato, documentato e convalidato.
6. Prima di ogni modifica significativa della lavorazione, il procedimento modificato è convalidato e documentato.
7. Le procedure di lavorazione sono periodicamente sottoposte a valutazione critica, per garantire la continuità del conseguimento dei risultati previsti.
8. Le procedure per scartare tessuti e cellule sono tali da impedire la contaminazione di altri tessuti e cellule, dell'ambiente di lavorazione o del personale. Tali procedure sono eseguite nel rispetto delle disposizioni vigenti.
C. Stoccaggio e rilascio di tessuti e cellule
Nel caso in cui le attività per le quali si chiede l'autorizzazione e l'accreditamento comprendano lo stoccaggio e il rilascio di tessuti e cellule, l'istituto dei tessuti si attiene alle procedure ed ai criteri seguenti:
1. Per ogni tipo di condizione di stoccaggio viene precisato il tempo massimo. Il periodo stabilito tiene conto, tra l'altro, dell'eventuale deterioramento delle proprietà richieste per tessuti e cellule.
2. Al fine di garantire che tessuti e cellule non siano rilasciati prima che siano state rispettate tutte le condizioni previste dal presente decreto, è necessario predisporre un inventario per i tessuti e/o le cellule; deve essere predisposta una procedura operativa standard che precisi le circostanze, le responsabilità nonchè le procedure inerenti al rilascio di tessuti e cellule per la distribuzione.
3. L'istituto dei tessuti pone in atto un sistema per l'identificazione di tessuti e cellule in ogni fase di lavorazione, mirato a distinguere in modo inequivocabile i prodotti rilasciati da quelli non rilasciati (in quarantena) e da quelli scartati.
4. I dati registrati sono in grado di dimostrare che prima del rilascio di tessuti e cellule sono state rispettate tutte le necessarie corrispondenti specifiche, in particolare che tutti i moduli di dichiarazione in uso, le cartelle mediche pertinenti, le registrazioni di lavorazione e i risultati dei controlli sono stati verificati in base a una procedura scritta da personale a tal fine autorizzato dal responsabile dell'istituto dei tessuti. Qualora i risultati di esami di laboratorio siano comunicati per via telematica, è indispensabile ne risulti traccia che permetta di individuare il responsabile del loro rilascio.
5. Ove siano introdotti nuovi criteri di selezione o controllo dei donatori o significative modifiche delle fasi di lavorazione per rafforzare la sicurezza o la qualità, è eseguita una valutazione dei rischi documentata, approvata dal responsabile dell'istituto dei tessuti, finalizzata a stabilire il destino di tutti i tessuti e le cellule stoccati.
D. Distribuzione e ritiro
Nel caso in cui le attività per le quali si chiede l'autorizzazione e l'accreditamento comprendano la distribuzione di tessuti e cellule, l'istituto dei tessuti si attiene alle procedure ed ai criteri seguenti.
1. Al fine di conservare le proprietà richieste per tessuti e cellule, sono definite le condizioni di trasporto critiche, quali: temperatura e scadenze temporali.
2. Il contenitore e l'imballaggio è realizzato in modo da risultare affidabile e garantire la conservazione di tessuti e cellule nelle condizioni prestabilite. Tutti i contenitori e gli imballaggi in uso sono convalidati come idonei allo scopo cui sono destinati.
3. Ove la distribuzione sia affidata a terzi, un accordo documentato garantisce il mantenimento delle prestabilite condizioni richieste.
4. Il personale dell'istituto dei tessuti, a tal fine incaricato, valuta le eventuali esigenze di ritiro ed avvia e coordina le azioni necessarie conseguenti.
5. L'istituto dei tessuti predispone una efficace procedura per il ritiro, che includa la descrizione delle responsabilità e delle azioni da intraprendere, compresa la notifica all'autorità competente.
6. Le azioni da intraprendere comunque entro un periodo predefinito comportano l'individuazione dei tessuti e cellule interessati e una ricostruzione del loro percorso. L'indagine ha lo scopo di: identificare ogni donatore che possa aver contribuito a causare la reazione nel ricevente, recuperare tessuti e cellule provenienti da detto donatore, informare destinatari e riceventi dei tessuti e cellule, prelevati dal medesimo, dell'eventuale rischio a cui possono essere esposti.
7. Le richieste di tessuti e cellule sono gestite secondo procedure prestabilite; le disposizioni seguite per l'assegnazione di tessuti e cellule a determinati pazienti o strutture sanitarie sono loro comunicate, se richiesto, e documentate.
8. E'predisposto un sistema documentato per il trattamento dei prodotti restituiti, comprendente, se del caso, i criteri per la loro iscrizione nell'inventario.
E. Etichettatura finale per la distribuzione [38]
1. Sul contenitore primario di tessuti o cellule viene indicato:
a) tipo di tessuti e cellule, numero d'identificazione o codice dei tessuti o cellule e, se del caso, numero del lotto o della partita;
b) identificazione dell'istituto dei tessuti;
c) data di scadenza;
d) in caso di donazione autologa, tale impiego è specificato mediante la dicitura: "esclusivamente per uso autologo", identificando donatore e ricevente;
e) in caso di donazioni con destinatario, l'etichetta indica il ricevente prescelto;
f) qualora tessuti e cellule risultino positivi a uno specifico marcatore di malattia infettiva, l'etichetta reca la dicitura: "RISCHIO BIOLOGICO";
g) codice unico europeo applicabile ai tessuti e alle cellule distribuiti a fini di applicazioni sull'uomo o sequenza d'identificazione della donazione applicabile ai tessuti e alle cellule rilasciati per la circolazione, non distribuiti a fini di applicazioni sull'uomo.
Se alcune delle informazioni di cui alle lettere d), e) e g) non possono essere incluse nell'etichetta del contenitore primario, sono tuttavia fornite su un foglio separato ad esso allegato. Tale foglio è imballato insieme al contenitore primario in modo da garantire che restino uniti.
2. Sull'etichetta o nella documentazione di accompagnamento sono riportate le seguenti informazioni:
a) descrizione e, se del caso, dimensioni del prodotto di tessuto o cellule;
b) morfologia e dati funzionali, se necessario;
c) data di distribuzione del tessuto o delle cellule;
d) analisi biologiche eseguiti sul donatore e risultati;
e) raccomandazioni per lo stoccaggio;
f) istruzioni per l'apertura del contenitore e dell'imballaggio e per ogni altra manipolazione o ricostituzione necessaria;
g) data di scadenza dall'apertura o manipolazione;
h) istruzioni per la notifica delle reazioni o degli eventi avversi gravi, di cui agli articoli 5 e 6;
i) presenza di residui potenzialmente nocivi (ad esempio antibiotici, ossido di etilene);
l) per i tessuti e le cellule importati, paese di approvvigionamento e paese esportatore (se differente dal paese di approvvigionamento).
F. Etichettatura esterna del contenitore per la spedizione
1. Ai fini del trasporto il contenitore primario è collocato per la spedizione in un contenitore, la cui etichetta reca almeno le seguenti informazioni:
a) identificazione dell'istituto dei tessuti d'origine, compresi indirizzo e numero telefonico;
b) identificazione dell'organizzazione responsabile dell'applicazione sull'uomo destinataria, compresi indirizzo e numero telefonico;
c) indicazione che l'imballaggio contiene tessuti e cellule umani con la dicitura:
"TRATTARE CON CAUTELA";
d) se ai fini del trapianto occorrono cellule viventi, quali cellule staminali, gameti e embrioni, è aggiunta la dicitura: "NON IRRADIARE";
e) condizioni di trasporto raccomandate (ad esempio conservare al fresco, in posizione verticale);
f) istruzioni per la sicurezza e metodo di raffreddamento, se necessario.
ALLEGATO X [40]
Informazioni sui dati minimi da conservare ai sensi dell'articolo 14
A. DA PARTE DEGLI ISTITUTI DEI TESSUTI
1) Identificazione del donatore
2) Identificazione della donazione che deve almeno comprendere:
- l'identificazione dell'organizzazione di approvvigionamento (compresi i dati di contatto) o dell'istituto dei tessuti,
- il numero unico della donazione,
- la data dell'approvvigionamento,
- il luogo dell'approvvigionamento,
- il tipo di donazione (ad esempio di un tessuto unico/di più tessuti, autologa/allogenica, da vivente/da deceduto).
3) Identificazione del prodotto che deve almeno comprendere:
- l'identificazione dell'istituto dei tessuti,
- il tipo di tessuti e cellule/prodotto (nomenclature di base),
- il numero aggregato delle partite (in caso di pooling),
- il numero specifico della sottopartita(se applicabile)
- la data di scadenza (se applicabile)
- lo stato dei tessuti/cellule (in quarantena, idonei all'uso ccc),
- la descrizione e l'origine dei prodotti, delle fasi di lavorazione in uso, dei materiali e degli additivi che vengono a contatto con tessuti e cellule e influiscono sulla loro qualità e/o sicurezza,
- l'identificazione del servizio responsabile dell'etichettatura finale.
4) Codice unico europeo (se applicabile)
5) Identificazione dell'applicazione sull'uomo che deve almeno comprendere:
- la data di distribuzione/smaltimento,
- l'identificazione del clinico o del servizio/utente finale.
B. DA PARTE DELLE ORGANIZZAZIONI RESPONSABILI DELL' APPLICAZIONE SULL'UOMO
1) Identificazione dell'istituto dei tessuti fornitore
2) Identificazione del clinico o del servizio/utente finale
3) Tipo di tessuti e cellule
4) Identificazione del prodotto
5) Identificazione del ricevente
6) Data dell'applicazione
7) Codice unico europeo (se applicabile)
ALLEGATO XI [41]
STRUTTURA DEL CODICE UNICO EUROPEO
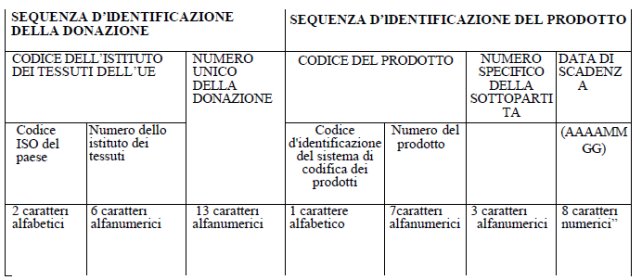
ALLEGATO XI-bis [42]
Dati da registrare nel compendio degli Istituti dei tessuti dell'UE
A. Informazioni sull'istituto dei tessuti
1. Nome dell'Istituto dei tessuti
2. Codice nazionale o internazionale dell'istituto dei tessuti
3. Nome dell'organizzazione presso la quale si trova l'istituto dei tessuti (se applicabile)
4. Indirizzo dell'Istituto dei tessuti
5. Dati di contatto pubblicabili: indirizzo di posta elettronica, telefono e fax.
B. Informazioni sull'autorizzazione, sull'accreditamento dell'istituto dei tessuti
1. Nome dell'autorità competente per l'autorizzazione, l'accreditamento
2. Nome dell'autorità competente a livello nazionale responsabili della gestione del compendio degli istituti dei tessuti dell'UE
3. Nome del titolare dell'autorizzazione, dell'accreditamento
4. Tessuti e cellule per i quali sono stati rilasciati l'autorizzazione e l'accreditamento
5. Attività effettivamente realizzate per le quali sono stati rilasciati l'autorizzazione, l'accreditamento
6. Stato dell'autorizzazione, dell'accreditamento (autorizzati, sospesi, revocati, in tutto o in parte, cessazione volontaria delle attività)
7. Informazioni su eventuali condizioni e deroghe aggiunte all'autorizzazione (se applicabile).
[1] Comma così modificato dall'art. 1 del
[2] Lettera aggiunta dall'art. 1 del
[3] Lettera aggiunta dall'art. 1 del
[4] Lettera aggiunta dall'art. 1 del
[5] Lettera aggiunta dall'art. 1 del
[6] Lettera aggiunta dall'art. 1 del
[7] Lettera aggiunta dall'art. 1 del
[8] Lettera aggiunta dall'art. 1 del
[9] Lettera aggiunta dall'art. 1 del
[10] Lettera aggiunta dall'art. 1 del
[11] Lettera aggiunta dall'art. 1 del
[12] Lettera aggiunta dall'art. 1 del
[13] Lettera aggiunta dall'art. 1 del
[14] Lettera aggiunta dall'art. 1 del
[15] Lettera aggiunta dall'art. 1 del
[16] Lettera aggiunta dall'art. 1 del
[17] Lettera aggiunta dall'art. 1 del
[18] Lettera aggiunta dall'art. 1 del
[19] Lettera aggiunta dall'art. 1 del
[20] Comma così modificato dall'art. 2 del
[21] Lettera così modificata dall'art. 3 del
[22] Lettera così modificata dall'art. 3 del
[23] Comma inserito dall'art. 3 del
[24] Lettera così modificata dall'art. 4 del
[25] Lettera così modificata dall'art. 4 del
[26] Lettera così modificata dall'art. 4 del
[27] Comma inserito dall'art. 4 del
[28] Articolo così sostituito dall'art. 2 del
[29] Articolo così sostituito dall'art. 3 del
[30] Articolo inserito dall'art. 4 del
[31] Articolo inserito dall'art. 4 del
[32] Articolo inserito dall'art. 5 del
[33] Punto così modificato dall'art. 1 del
[34] Paragrafo già modificato dall'art. 6 del
[35] Punto sostituito dall'art. 6 del
[36] Lettera così modificata dall'art. 7 del
[37] Punto così modificato dall'art. 8 del
[38] Parte così modificata dall'art. 6 del
[39] Gli allegati VII e VIII sono stati sostituiti dall'art. 6 del
[40] Allegato così sostituito dall'art. 6 del
[41] Allegato così sostituito dall'art. 6 del
[42] Allegato inserito dall'art. 6 del